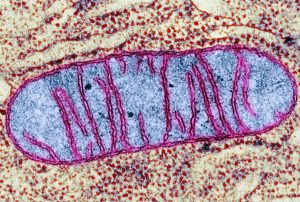
میتوکندری، اندامک حیاتی سلول، مسئول تولید حدود ۹۰٪ انرژی موردنیاز سلولها در قالب آدنوزین تریفسفات (ATP) از طریق فرآیند اکسیداتیو فسفوریلاسیون (OXPHOS) است. این اندامک نهتنها در تولید انرژی نقش دارد، بلکه در مسیرهای سیگنالدهی مانند چرخه اسید تریکربوکسیلیک (TCA)، β-اکسیداسیون اسیدهای چرب، تنظیم کلسیم و کنترل آپوپتوز دخیل است. در این مقاله، به بررسی ساختار DNA میتوکندریایی (mtDNA)، نقش آن در عملکرد سلولی، جهشهای مرتبط با بیماریها و مکانیزمهای انتقال آن میپردازیم.
میتوکندری: نیروگاه سلول
میتوکندریها با تولید ATP از طریق OXPHOS، انرژی لازم برای فعالیتهای سلولی را تأمین میکنند. این اندامک دو غشایی همچنین در تنظیم فرآیندهای کلیدی مانند چرخه سلولی و آپوپتوز (مرگ برنامهریزیشده سلول) نقش دارد. برخلاف سایر اندامکها، میتوکندری دارای DNA اختصاصی (mtDNA) است که ژنهای حیاتی برای تنفس سلولی را کد میکند.
ساختار DNA میتوکندریایی (mtDNA)
mtDNA برخلاف DNA هستهای (nDNA)، مشابه کروموزومهای باکتریایی، بهصورت دایرهای دو رشتهای است و در ماتریکس میتوکندری قرار دارد. در انسان، mtDNA شامل ۱۶۵۶۹ جفت باز و ۳۷ ژن است که شامل:
- ۱۳ پلیپپتید: زیرواحدهای اصلی کمپلکسهای OXPHOS (I، III، IV و V).
- ۲ RNA ریبوزومی و ۲۲ tRNA: برای ترجمه پروتئینهای کدشده.
- حلقه جابهجایی (D-loop): منطقه غیرکدکننده که نقاط آغاز تکثیر و رونویسی را دربرمیگیرد.
رشتههای mtDNA به نامهای سنگین (H) و سبک (L) شناخته میشوند، که ژنهای متفاوتی را کد میکنند. این ساختار توسط پروتئینهایی مانند TFAM، POLG و پروهیبیتینها بهصورت نوکلئوئید بستهبندی میشود. TFAM نقش کلیدی در رونویسی و سازماندهی mtDNA دارد.
جهشهای mtDNA و بیماریهای انسانی
mtDNA به دلیل فقدان هیستونها و مکانیزمهای تعمیر محدود، نسبت به آسیبهای ناشی از گونههای فعال اکسیژن (ROS) بسیار حساس است. این آسیبها میتوانند جهشهای سوماتیک ایجاد کنند که عملکرد OXPHOS را مختل کرده و به بیماریهای مختلفی منجر شوند. بیماریهای مرتبط با mtDNA اغلب علائم نورولوژیکی دارند و به دلیل وراثت مادری mtDNA، از مادر به فرزندان منتقل میشوند.
نمونههایی از بیماریهای مرتبط با mtDNA
از زمان گزارش اولین جهش mtDNA در سال ۱۹۸۸، چندین بیماری مرتبط با جهشهای mtDNA شناسایی شدهاند، از جمله:
- سندرمهای نورولوژیکی: مانند نوروپاتی، آتاکسی و صرع.
- اختلالات متابولیکی: ناشی از نقص در تولید ATP.
- بیماریهای میتوکندریایی: مانند سندرم لی (Leigh) و MELAS.
میتوکندریهای آسیبدیده معمولاً از طریق میتوفاژی، فرآیندی که توسط پروتئینهای PINK1 و Parkin هدایت میشود، حذف میشوند.
وراثت مادری mtDNA
mtDNA بهطور انحصاری از مادر به ارث میرسد و mtDNA پدری در هنگام لقاح حذف میشود. مکانیزم حذف mtDNA پدری، که شامل میتوفاژی و اندونوکلئاز G است، یکی از موضوعات مورد توجه در زیستشناسی سلولی است. درک این فرآیند میتواند به فهم بهتر بیماریهای میتوکندریایی کمک کند.
نقش پروتئینهای نوکلئوئید
پروتئینهایی مانند TFAM، ATAD3 و POLG در بستهبندی و توزیع mtDNA نقش دارند. نقص در توزیع mtDNA میتواند عملکرد میتوکندری را مختل کرده و به بیماریهای مرتبط با انرژی سلولی منجر شود. برای مثال، TFAM بهعنوان فاکتور اصلی رونویسی، در حفظ ساختار و عملکرد mtDNA حیاتی است.
عوامل محیطی و سلامت میتوکندری
عوامل محیطی مانند استرس اکسیداتیو، تغذیه نامناسب و کمبود خواب میتوانند به mtDNA آسیب برسانند. سبک زندگی سالم، از جمله رژیم غذایی متعادل و ورزش منظم، میتواند به حفظ سلامت میتوکندری و کاهش خطر جهشهای mtDNA کمک کند.
فناوری و آینده پژوهشهای میتوکندری
پیشرفتهای اخیر در فناوریهایی مانند ویرایش ژن (CRISPR) و تصویربرداری پیشرفته، امکان مطالعه دقیقتر mtDNA و مکانیزمهای آن را فراهم کرده است. این ابزارها میتوانند به توسعه درمانهایی برای بیماریهای میتوکندریایی کمک کنند.
نتیجهگیری: میتوکندری، کلید حیات و بیماری
میتوکندریها فراتر از تولید انرژی، در تنظیم فرآیندهای حیاتی سلولی نقش دارند. mtDNA به دلیل ساختار منحصربهفرد و حساسیت به جهش، در سلامت و بیماریهای انسانی اهمیت زیادی دارد. با درک بهتر مکانیزمهای mtDNA و تأثیر عوامل محیطی، میتوان راهکارهایی برای پیشگیری و درمان بیماریهای مرتبط با میتوکندری توسعه داد.
 از
از